Three classes of propofol binding sites on GABAA receptors.
Study Design
- Loại nghiên cứu
- Other
- Đối tượng nghiên cứu
- None
- Can thiệp
- Three classes of propofol binding sites on GABAA receptors. 3 G
- Đối chứng
- None
- Kết quả chính
- None
- Xu hướng hiệu quả
- Neutral
- Nguy cơ sai lệch
- Unclear
Abstract
Propofol is a widely used anesthetic and sedative that acts as a positive allosteric modulator of gamma-aminobutyric acid type A (GABAA) receptors. Several potential propofol binding sites that may mediate this effect have been identified using propofol-analogue photoaffinity labeling. Ortho-propofol diazirine (o-PD) labels β-H267, a pore-lining residue, whereas AziPm labels residues β-M286, β-M227, and α-I239 in the two membrane-facing interfaces [β(+)/α(-) and α(+)/β(-)] between α and β subunits. This study used photoaffinity labeling of α1β3 GABAA receptors to reconcile the apparently conflicting results obtained with AziPm and o-PD labeling, focusing on whether β3-H267 identifies specific propofol binding site(s). The results show that propofol, but not AziPm protects β3-H267 from labeling by o-PD, whereas both propofol and o-PD protect against AziPm labeling of β3-M286, β3-M227, and α1I239. These data indicate that there are three distinct classes of propofol binding sites, with AziPm binding to two of the classes and o-PD to all three. Analysis of binding stoichiometry using native mass spectrometry in β3 homomeric receptors, demonstrated a minimum of five AziPm labeled residues and three o-PD labeled residues per pentamer, suggesting that there are two distinct propofol binding sites per β-subunit. The native mass spectrometry data, coupled with photolabeling performed in the presence of zinc, indicate that the binding site(s) identified by o-PD are adjacent to, but not within the channel pore, since the pore at the 17' H267 residue can accommodate only one propofol molecule. These data validate the existence of three classes of specific propofol binding sites on α1β3 GABAA receptors.
Tóm lược
The results show that propofol, but not AziPm protects β3-H267 from labeling by o-PD, whereas both propofol and o-PD protect against AziPm labeling of β3-M286, β3-M227, and α1I239, and validate the existence of three classes of specific propofol binding sites on α1β3 GABAA receptors.
Full Text
RESEARCH ARTICLE
Three classes of propofol binding sites on GABAA receptors
Received for publication, January 9, 2024, and in revised form, September 4, 2024 Published, Papers in Press, September 11, 2024, https://doi.org/10.1016/j.jbc.2024.107778
Zi-Wei Chen1,2, Satyanarayana M. Chintala1, John Bracamontes1, Yusuke Sugasawa1,3 , Spencer R. Pierce1, Balazs R. Varga1, Edward H. Smith4, Christopher J. Edge4, Nicholas P. Franks4,5, Wayland W. L. Cheng1, Gustav Akk1,2, and Alex S. Evers1,6,2,*
From the 1Department of Anesthesiology, Washington University School of Medicine, St Louis, Missouri, USA; 2The Taylor Family Institute for Innovative Psychiatric Research Washington University School of Medicine, St Louis, Missouri, USA; 3Department of Anesthesiology and Pain Medicine, Juntendo University School of Medicine, Tokyo, Japan; 4Department of Life Sciences, Imperial College, London, UK; 5UK Dementia Research Institute, Imperial College, London, UK; 6Department of Developmental Biology, Washington University School of Medicine, St Louis, Missouri, USA
Reviewed by members of the JBC Editorial Board. Edited by Mike Shipston
subunit markedly attenuate propofol anesthesia in mice (2). Propofol can both potentiate currents elicited by low concentrations of GABA and directly activate GABAA currents (3, 4) and functional analysis using the Monod–Changeux– Wyman model indicates that both of these effects are mediated by the same set of propofol binding sites (5).
Propofol is a widely used anesthetic and sedative that acts as a positive allosteric modulator of gamma-aminobutyric acid type A (GABAA) receptors. Several potential propofol binding sites that may mediate this effect have been identified using propofol-analogue photoaffinity labeling. Ortho-propofol diazirine (o-PD) labels b-H267, a pore-lining residue, whereas AziPm labels residues b-M286, b-M227, and a-I239 in the two membrane-facing interfaces [b(+)/a(−) and a(+)/b(−)] between
GABAA receptors are pentameric ion channels composed of varying combinations of 19 subunits (6a, 3b, 3g, 3r, 1d, 1ε, 1p, and 1q) each composed of an extracellular domain, a transmembrane domain (TMD) and an intracellular domain. The subunits are arranged in a concentric circle creating an internal ion conducting pore. The TMDs of each subunit are composed of four membrane spanning a-helices (TM1-TM4), with TM2 of each subunit lining the pore and the subunits interacting by contact between TM3 of one subunit (designated the “+” side) and TM1 of an adjacent subunit (designated the “-” side) (6–8).
- a and b subunits. This study used photoaffinity labeling of a1b3 GABAA receptors to reconcile the apparently conflicting results obtained with AziPm and o-PD labeling, focusing on whether b3-H267 identifies specific propofol binding site(s). The results show that propofol, but not AziPm protects b3-H267 from labeling by o-PD, whereas both propofol and o-PD protect
against AziPm labeling of b3-M286, b3-M227, and a1I239. These data indicate that there are three distinct classes of propofol binding sites, with AziPm binding to two of the classes and o-PD to all three. Analysis of binding stoichiometry using native mass spectrometry in b3 homomeric receptors, demonstrated a minimum of five AziPm labeled residues and three o-PD labeled residues per pentamer, suggesting that there are two distinct propofol binding sites per b-subunit. The native mass spectrometry data, coupled with photolabeling performed in the presence of zinc, indicate that the binding site(s) identified by o-PD are adjacent to, but not within the channel pore, since the pore at the 170 H267 residue can accommodate only one propofol molecule. These data validate the existence of three classes of specific propofol binding sites on a1b3 GABAA receptors.
Binding sites for propofol on GABAA receptors have been identified using photoaffinity labeling with subsequent protein sequencing by either mass spectrometry (MS) or Edman degradation. The b3-H267 residue (170 on TM2), is labeled by ortho-propofol diazirine (o-PD; Fig. 1F) in b3-homomeric and a1b3 GABAA receptors (9). Molecular docking studies indicate a putative propofol binding pocket between TM2 and TM1 of one b3 subunit and TM2 of an adjacent subunit (Fig. 1) (10). Based on the location of H267, this binding pocket is referred to as a pore-adjacent site. The functional effects of o-PD on GABAA receptor currents closely replicate those of propofol (9). However, the photochemical products of o-PD preferentially label nucleophilic amino acids such as histidine and may fail to label other residues with which it is proximal. Metaazipropofol (AziPm; Fig. 1G) labels a distinct set of residues in a1b3 and a1b3g2 GABAA receptors (b3-M286 on TM3, b3M227 on TM1 and a1-I239 on TM1) indicating a binding site in the b3(+)/a1(−) subunit interface and likely binding sites in the a1(+)/b3(−) and/or g2(+)/b3(−) subunit interfaces (Fig. 1) (11). These sites face the lipid bilayer and are referred to as membrane-adjacent sites. AziPm is an excellent photolabeling reagent with no preference for specific amino acids, but poorly
The gamma-aminobutyric acid type A (GABAA) receptor is considered to be the major molecular target responsible for the anesthetic effect of propofol, the most widely used intravenous anesthetic and sedative in the world. This is supported by the strict correlation of anesthetic activity of propofol analogues with their ability to modulate GABAA receptor currents (1) and by the observation that point mutations in the GABAA-b3
* For correspondence: Alex S. Evers, [email protected]. replicates the actions of propofol on GABAA receptors: it
J. Biol. Chem. (2024) 300(10) 107778 1
© 2024 THE AUTHORS. Published by Elsevier Inc on behalf of American Society for Biochemistry and Molecular Biology. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/).
- Figure 1. Propofol binding sites predicted by photolabeling on an a1b3 GABAA receptor. A, side view of an a1b3 GABAA receptor (PDB: 7PBD) with transmembrane domain helices illustrated as cylinders. The a1 subunits are shown in yellow and the b3 subunit in cyan. Propofol molecules are illustrated as ovals with the orange propofol in the a1(+)/b3(−) membrane-adjacent site, the purple propofol in the pore-adjacent site and the salmon propofol in the b3(+)/a1(−) membrane-adjacent site. B, magnified side view of binding sites viewed from the membrane face of the receptor with propofol molecules docked in their energetically preferred pose in each binding site. The membrane-adjacent sites are approximately two a-helical turns below the pore-
adjacent site. Residues photolabeled by propofol analogues are shown in stick format. C, vertical view of the a1b3 GABAA receptor from the extracellular side illustrating that the putative membrane-adjacent propofol binding sites are on the outside of the receptor while the putative pore-adjacent sites are on the pore-facing side. D, magnified extracellular vertical view with propofol molecules docked in their energetically preferred pose in each site. E–H, molecular structures of propofol and the photolabeling analogues o-PD, AziPm, and AziPm-d9. GABAA, gamma-aminobutyric acid type A; o-PD, orthopropofol diazirine; PDB, Protein Data Bank.
weakly potentiates GABA-elicited currents and does not directly activate the channel (12).
Site-directed mutagenesis studies provide strong evidence that propofol binding to both the pore- and membraneadjacent sites predicted by photolabeling are involved in the action of propofol. Tryptophan mutations in the pore-adjacent site (Y143W, F221W, and Q224W) of b3-homomeric GABAA receptors markedly reduce direct activation by propofol (13). In a1b3 receptors, tryptophan mutations in the pore- or membrane-adjacent b3(+)/a1(−) sites (M286W) reduce propofol potentiation of GABA-elicited currents but do not reduce direct activation. In contrast, double mutations (in both the pore- and membrane-adjacent sites) eliminate potentiation and strongly reduce activation (14). Similar effects of the M286W mutation have been observed with a1b2g2 receptors (15, 16).
Despite the photolabeling and mutagenesis data, there has been skepticism regarding the existence and functional importance of the pore-adjacent binding site, with recent
literature focusing on the b3(+)-a1(−) intersubunit site as the sole or dominant site for propofol binding and effect (17, 18). Skepticism about the pore-adjacent site arises from the absence of evidence that excess propofol prevents o-PD labeling (9, 11) and from the minimal effect of H267 mutations on propofol action (13, 19). Support for the view that the b3(+)/a1(−) site is the sole site is enhanced by the demonstration of a propofol density in this site, but not in the pore-adjacent or membraneadjacent a1(+)/b3(−) sites, in a cryogenic electron microscopy density map of the a1b2g2 receptor (17). The current study revisits propofol binding sites, providing additional photolabeling data and placing photolabeling and mutagenesis studies in the context of more recent structural studies. We provide new evidence for the existence and specificity of the pore-adjacent site and show that o-PD binds to both the poreand membrane-adjacent sites. o-PD does not label the membrane-adjacent sites, likely due to its photochemical specificity. The data also demonstrate that AziPm does not
bind to the pore-adjacent site and thus labels only the membrane-adjacent propofol sites. This provides a potential explanation for the inability of AziPm to mimic the effects of propofol on GABAA receptor gating.
Results
Prevention of o-PD labeling by propofol and AziPm
To determine whether the b-H267 residue labeled by o-PD identified a specific binding site, we examined the ability of propofol to prevent o-PD labeling. To perform this analysis, we photolabeled HEK cell membranes containing a1FLAG-Hisb3GABAA receptors, solubilized the receptors in n-dodecyl-betamaltoside (DDM) and affinity-enriched the GABAA receptors using the FLAG epitope tag (20). We then digested the protein with trypsin and directly analyzed the resulting peptides by LC-MS (without solid phase extraction). This allowed us to obtain the full-length tryptic a-helical (TM1-4) peptides from the a1 and b3 subunits, enabling direct comparison of the intensity of the labeled and unlabeled peptides. In our previous analyses (9, 21), we used multiple, timed chymotryptic and tryptic digests to sequence the receptor. This produced multiple overlapping peptides with cleavages influenced by the presence of the o-PD adduct, making it difficult to assess the relative abundance of the unlabeled and photolabeled peptide.
Using this approach, we identified all four of the TM peptides from both the a1 and b3 subunits of the GABAA receptor with 100% sequence coverage of the TMDs. Two photolabeled peptides were identified: the tryptic TM2 peptide 251VALGITTVLTMTTINTHOPDLR269 (m/z = 757.77; z = 3) and the same peptide with a single missed cleavage, 251VALGITTVLTMTTINTHOPDLRETLPK274 (m/z = 947.191, z = 3 and m/z = 710.65, z = 4), each bearing a single o-PD adduct (mass = 216.08). These peptides met our predetermined criteria for a photolabeled peptide: (1) clearly defined charge states; (2) mass accuracy <20 ppm from theoretical m/z; (3)
delayed chromatographic retention time ( 2 min) compared to the unlabeled peptide (effect of the hydrophobic adduct); and (4) site-defining fragment ions for the adduct with mass accuracy <20 ppm. The fragment ion spectrum of 251VALGITTVLTMTTINTHOPDLRETLPK274, displayed a series of y ions containing the o-PD adduct (y8-y13, marked in red; mass = 216.08) and a series of y ions (y1–y7) not containing the o-PD adduct (Fig. 2). This spectrum defines the o-PD labeled residue as H267.
Photolabeling with o-PD (10 mM) was then conducted in the presence and absence of 300 mM propofol. The mean labeling efficiency of the TM2 peptides was 0.70 ± 0.07% in control conditions and 0.10 ± 0.08% in the presence of propofol (n = 3; p < 0.01; Fig. 2B). These data indicate that excess propofol prevents o-PD photolabeling of b3-H267, consistent with competitive inhibition of specific binding. We next sought to determine whether AziPm prevents o-PD labeling of H267. AziPm and o-PD have the same molecular mass rendering their photo-adducts indistinguishable by mass spectrometry. To assess whether AziPm prevents o-PD labeling we synthesized the deuterated analogue, AziPm-d (Fig. 1H; n.b. AziPmd is a mixture of the d6, d7, d8, and d9 analogues). a1b3 GABAA receptors were labeled with 10 mM o-PD in the presence and absence of 300 mM of AziPm-d. AziPm-d had no significant effect on o-PD labeling of TM2-H267 (Fig. 2B). Even at 300 mM, no AziPm-d-labeled TM2 peptides were identified. However, AziPm-d (300 mM) did label the same peptides/ residues in the membrane adjacent sites that are labeled by AziPm (see below). These data indicate that AziPm-d is an effective photolabeling reagent, but does not prevent labeling of the specific binding site identified by o-PD.
Prevention of AziPm-d labeling by propofol and o-PD
A previous study of AziPm photolabeling of purified a1b3 GABAA receptors using Edman degradation for sequencing
- Figure 2. o-PD photolabeling of the H267 residue on b3-TM2 of an a1b3 GABAA receptor is inhibited by propofol, but not AziPm-d. A, fragment ion spectrum of o-PD (10 mM) photolabeling of H267 on the b3-TM2 peptide 251VALGITTVLTMTTINTHLRETLPK274. The y8*-y13* fragment ions (red*) contain the o-PD adduct, whereas the y6 and y7 fragment ions (black) do not, indicating H267 as the photolabeled residue. The unlabeled (black) y8 and y9 ions represent neutral loss. B, photolabeling of the b3-TM2 peptide by o-PD (10 mM) in the presence and absence of propofol (300 mM) or AziPm-d (300 mM). Propofol but not AziPm-d prevented o-PD photolabeling. Results were compared using a two-tailed paired t test. ** = p < 0.01. GABAA, gammaaminobutyric acid type A; o-PD, ortho-propofol diazirine; TM, transmembrane.
identified three labeled residues: b3-TM3-M286, b3-TM1M227, and a1-TM1-I239. Photolabeling of all of these peptides was prevented by an excess of propofol (11). We repeated this analysis, photolabeling a1b3 GABAA receptors with 10 mM AziPm and identifying the labeled peptides/ residues using LC-MS as described above. Three GABAA
receptor tryptic peptides were observed with AziPm adducts (MW = 216.08), all meeting our predetermined criteria for a photolabeled peptide: (1) A b3-TM3 peptide, 280AIDMYLMAziPmGCNEMFVFVFLALLEYAFVNYIFFGR313 (m/z = 1305.29, z = 3), with an N-ethylmaleimide (NEM) adduct on C288 and an AziPm adduct on M286 (Fig. 3A).
- Figure 3. AziPm photolabeling of membrane-adjacent sites in a1b3 GABAA receptors is inhibited by both propofol and o-PD. A, fragment ion spectrum of b3-TM3 peptide photolabeled by AziPm on M286 as identified by site defining ions b6 and b7* (fragment ions containing the adduct are shown in red with a star; ions not containing the adduct are shown in black). B, fragment ion spectrum of a1-TM1 photolabeled by AziPm on I239 as identified by site defining ions y10 and y11*. C, overlay of the fragment ion spectra of b3-TM1 peptide photolabeled by AziPm and AziPm-d7 on M227 as identified by site defining ions b10 and b11*. The inset illustrates that fragment ions not containing the adduct (b10/b10) are identical in the AziPm and AziPm-d7 spectra, whereas fragment ions containing the adduct (b11AziPm/b11AziPm-d7) differ by 7 da. Precursor ions with an AziPm-d8 and AziPm-d9 adduct were also detected; in fragmentation spectra of these precursors, fragment ions containing the adduct differed by 8 or 9 mass units from the AziPm adduct, respectively. D, photolabeling of b3-TM3, a1-TM1, and b3-TM1 peptides by AziPm is reduced in the presence of propofol (1 mM). E, photolabeling of b3-TM3, a1-TM1, and b3-TM1 peptides by AziPm-d is reduced in the presence of o-PD (300 mM). Data in panels D and E were analyzed using a paired t test. * = p < 0.05. GABAA, gamma-aminobutyric acid type A; o-PD, ortho-propofol diazirine; TM, transmembrane.
The photolabeling efficiency was 0.53 ± 0.15% (mean ± SEM), which was reduced to 0.08 ± 0.09% (p < 0.05; n = 3) in the presence of 1 mM propofol (Fig. 3D); (2) an a1-TM1 peptide 223IGYFVIQTYLPCMTVIAziPmLSQVSFWLNR249 (m/z = 1140.959, z = 3) with an AziPm adduct on I239 (Fig. 3B). The photolabeling efficiency was 3.80 ± 0.69% which was reduced to 0.47 ± 0.44% in the presence of 1 mM propofol (p < 0.01; n = 3) (Fig. 3D); and (3) a b3-TM1 peptide, 217NIGYFILQTYMAziPmPSILITILSWVSFWINYDASAAR250 (m/z = 1046.583, z = 4) with an AziPm adduct on M227 (Fig. 3C). The photolabeling efficiency was 3.70 ± 0.08% which was reduced to 0.37 ± 0.02% in the presence of 1 mM propofol (p < 0.01; n = 3) (Fig. 3D). No photolabeling of b3-TM2 was observed. These results replicate the AziPm labeled peptides and residues identified using Edman degradation and validate the comparability of the methods.
To investigate whether o-PD prevents AziPm binding, we labeled a1b3 GABAA receptors with 10 mM AziPm-d in the presence and absence of 300 mM o-PD. AziPm-d labeled the same three peptides and localized to the same residues as AziPm with similar labeling efficiency (Fig. 3E). Overlay of the fragment ion spectra from the AziPm and AziPm-d labeled peptides provided additional data for adduct site localization and confirmed that both ligands formed adducts with the same residues. This is illustrated for the AziPM and AziPm-d7 labeled b3-TM3 peptide, 280AIDMYLMAziPmGCNEMFVFVFLALLEYAFVNYIFFGR313 (Fig. 3C) in which fragment ions containing the AziPm adduct (e.g. b11) differ by seven mass units between the heavy (AziPm-d7) and light AziPM, whereas fragment ions not containing the adduct (e.g. b10) have identical m/z. (n.b. TM2 peptides containing AziPm-d6, -d8, and d9-labeled peptides were also detected and the fragment ions containing the adduct differed from the AziPm-containing fragment ions by 6, 8, and 9 mass units, respectively.) Both propofol (1 mM) and o-PD (300 mM) significantly reduced AziPm photolabeling efficiency of all three labeled peptides
- (Fig. 3, D and E). These data indicate that o-PD prevents AziPm labeling, consistent with o-PD binding to these sites.
Modulation of GABAA receptor currents by o-PD, AziPm, and propofol
To explore the relationship between differential binding of oPD and AziPm to pore-adjacent versus membrane-adjacent propofol binding sites and their ability to modulate GABAA receptors,wecomparedthe ability of propofol,o-PD,and AziPm to potentiate GABA-elicited currents and to directly activate currentsina1b3GABAAreceptorsexpressedinXenopusoocytes.For theseexperiments,currentsareexpressedasthepercentage(%)of the response to a saturating (30 mM) concentration of GABA.
To assess the ability of the propofol analogues to directly
activate GABAA receptors, the receptors were exposed to high concentrations (100 mM) of propofol, o-PD, and AziPm. Pro-
pofol and o-PD produced strong direct activation (Po values of 92 ± 17% and 88 ± 4%, respectively), whereas AziPm showed
minimal direct activation (Po 1%) (Table 1). To assess the
ability of the propofol analogues to potentiate the effects of GABA, GABA was first applied at concentrations that produced 7 to 9% Po, followed by application of 1, 10, or 50 mM concentrations of propofol, o-PD, and AziPm. Propofol and
- o-PD strongly potentiated GABA-elicited currents (maximal Po values of 95 ± 7% and 92 ± 2%, respectively) with apparent EC50 values between 1 and 10 mM (Table 1). In contrast, AziPm potentiated GABA-elicited currents less potently and
- o-PD, and AziPm in the three sites identified by photolabeling, we performed in vacuo docking to the cryo-EM structure of an
- o-PD are identically oriented in a cavity defined by b3-Y143, F221, and Q224 (in yellow), adjacent to the TM2 domain of a1 at the a1(+)/b3(−) interface. These amino acids have been previously identified as contributing to propofol potentiation
- of GABAA currents in site-directed mutagenesis studies using electrophysiological readout (13). The diazirine group of o-PD is situated either 9.2 Å or 8.4 Å from the s-nitrogen on the imidazole ring of H267 (depending on the orientation of the ortho substituents of o-PD in the binding site), suggesting that H267 may not directly interact with propofol in the binding cavity. Unlike propofol and o-PD, AziPm did not dock in this site. When we attempted to position AziPm in the binding pocket with its hydroxyl group and isopropyl groups in the same position as propofol, the meta-TPD group of AziPm
sterically clashes with either b3-F221 on TM1 or b3-I281 on TM3, depending on which of the propofol isopropyl groups AziPm is aligned with. This provides a plausible explanation for the inability of AziPm to bind in the pore-adjacent site.
In the membrane-adjacent b3(+)/a1(−) interface site, the most energetically favorable poses for propofol, o-PD and AziPm overlap in position and orientation (Fig. 4C). The ligands assume an orientation similar to that observed for propofol in an a1b3g2 GABAA receptor cryo-EM structure (PDB: 6X3T) (17), with the aromatic ring of propofol facing the M286 side chain and the ortho and meta substituents
Table 1 Direct activation and potentiation of GABA-elicited currents in a1b3 GABAA receptors by propofol analogues
Low GABA + 50 mM
Ligand Direct activation by 100 mM Low GABA (0.1–0.3 mM) Low GABA + 1 mM Low GABA + 10 mM
Propofol 92 ± 17% 9 ± 4% 22 ± 12% 78 ± 15% 95 ± 7% AziPm 1.03 ± 0.95% 7 ± 2% 6 ± 1% 14 ± 7% 33 ± 14% o-PD 88 ± 4% 7 ± 3% 31 ± 9% 85 ± 6% 92 ± 2%
The a1b3 GABAA receptors were activated with the indicated concentrations of propofol, AziPm, and o-PD in the presence or absence of GABA. Currents are expressed as the percentage (%) of the response to a saturating concentration (30 mM) GABA. n = 5 for each condition.
arranged vertically in the b3(+)/a1(−) interface. The diazirine moieties of o-PD and AziPm and one of the isopropyl groups of propofol are positioned just above the side chain of M236. The diazirine groups of both o-PD and AziPm are within 6 Å of the backbone a-carbon of b3-M286 and a1-M236. a1-I239, the residue photolabeled with the highest efficiency by AziPm
- (Fig. 3, E and F), is oriented toward the same interface as
b3-M236 but positioned one a-helical turn below M236; the I239 backbone is positioned 11 Å away from the docked position of the AziPm diazirine group.
In the a1(+)/b3(−) membrane-adjacent site (near b3-M227), the energetically preferred poses for propofol, o-PD, and
AziPm overlap closely in their position in the interface (Fig. 4D). The phenyl groups of all three ligands are parallel to a1-TM3 and b3-TM1 helices with the hydroxyl group pointed into the protein toward TM2. The ortho and meta side chains are oriented vertically in the interface with the diazirine groups of o-PD and AziPm oriented in opposite directions. The diazirine group of AziPm is positioned 6 Å away from the backbone a-carbon of b3-M227. These docking results support the finding that o-PD binds to the two membrane-adjacent propofol binding sites labeled by AziPm. When an openstate structure becomes available, molecular dynamic simulations of propofol, o-PD, and AziPm binding to an open-state
- Figure 4. Computational docking of propofol, o-PD, and AziPM in the pore-adjacent and membrane-adjacent sites in an a1b3 GABAA cryogenic electron microscopy structure (PDB: 7PBD). A, top-down view (from an extracellular perspective) showing the most energetically favorable poses of
propofol (pink) and o-PD (blue) docked in the pore-adjacent site. The a1 subunit is shown in beige and the b3 subunit in cyan. Residues b3-Y143, -F221, and -Q224 in the binding pocket are shown in yellow stick and ball format; the H267 residue photolabeled by o-PD is shown in purple. AziPm is not shown because it did not dock in this site. B, side view (from membrane perspective) of propofol and o-PD docked in the pore-adjacent site. C and D, side views (from the membrane perspective) of propofol (pink), o-PD (blue), and AziPm (orange) docked in their most energetically favorable poses in the: (C) b3(+)/ a1(−) and; (D) a1(+)/b3(−) membrane-adjacent sites. Photolabeled residues are shown in stick and ball format. GABAA, gamma-aminobutyric acid type A; oPD, ortho-propofol diazirine; PDB, Protein Data Bank.
structure should provide more definitive information regarding the molecular details of propofol (and propofol analogue) binding to all of its sites on the GABAA receptor.
Stoichiometry of o-PD and AziPm labeling of b3-homomeric GABAA receptors determined by native mass spectrometry
b3-H267 is located at the 170 position in the pore of the GABAA receptor and its imidazole side chain can be rotated either into the pore, or toward the adjacent TM2 along the wall of the pore (22). In principle, o-PD could label H267 either from the pore or by entering a pore-adjacent binding site and accessing H267 from within this pocket. Since the diameter of the pore at the 170 position is 9 Å and the crosssectional area of a propofol molecule is 8.5 Å, the pore can only accommodate a single propofol and a photolabeling reagent in the pore could only label a single H267 per pentamer. If o-PD accesses H267 from a pocket located in each subunit, it could in principle label all of the H267 residues in a GABAA pentamer (2–5 depending on the number of b-subunits in the pentamer). To determine the stoichiometry of o-PD and AziPm photolabeling of a GABAA receptor, we repeatedly labeled the receptors with high concentrations of photolabeling reagent (100 mM X 3 for o-PD, 200 mM X 3 for AziPm) and analyzed the number of covalent adducts on the pentamer using native mass spectrometry. We used purified b3-homomeric receptors for these experiments to maximize the potential number of pore-adjacent sites. The spectra for the unlabeled homomeric b3-GABAA receptors (upper panels Fig. 5, A and C) show a single species with narrow charge state distribution (+27 to +29 and + 29 to +31) corresponding to an average mass of 215 kDa. This mass is consistent with a b3-GABAA receptor, which includes the calculated mass of the b3-ICL-del subunit construct (40.38 X 5 = 201.93 kDa) plus the weight of three Man5-GlcNac2 glycans per subunit obtained in GNTI- cells ( 13.5 kDa) (23). The spectrum of the AziPm-labeled receptors had a significantly broader feature for each charge state (Fig. 5A, lower panel) than the unlabeled receptor. Examination of the +28-charge state (the highest intensity charge state) shows a peak corresponding to the unlabeled pentamer plus five more peaks each with an average additional mass of 216 ± 17 Da (shown in red in Fig. 5B), consistent with the calculated adduct mass (216 Da) of AziPm. This result indicates a labeling stoichiometry of at least five sites per pentameric receptor. A spectrum of o-PD-labeled b3 homomeric receptors also showed a broader feature than the unlabeled receptor (Fig. 5C, lower versus upper panel). This spectrum showed lower signal-to-noise than the AziPm spectra, possibly due to an effect of the excess o-PD ligand on electrospray ionization. Nevertheless, the +30-charge state (the highest intensity charge state) shows a peak corresponding to the mass of the unlabeled receptor plus three more peaks with an average additional mass of 232 ± 42, consistent with the theoretical o-PD adduct mass (216 Da). These data indicate a minimum o-PD labeling stoichiometry of three sites per pentamer. The labeling stoichiometry of three per pentamer is likely a reflection of the maximum labeling efficiency that can
be achieved, limited by o-PD concentration, rather than the maximum binding stoichiometry.
Zinc inhibition of o-PD photolabeling of b3-H267
To confirm that o-PD binds in multiple pore-adjacent propofol binding sites, we exploited the observation that zinc (Zn2+) acts as a channel blocker for a1b3 and b3 homomeric GABAA receptors by forming a ternary complex with three H267 molecules at the 170 position in the pore (24). H267 imidazole side chains complexed to Zn2+ in the center of the pore should be inaccessible to labeling by o-PD located in pore-adjacent propofol binding pockets. Zn2+ should thus prevent labeling of Zn2+-complexed H267 residues. For a1b3 receptors, all three of the b3-H267 residues are coordinated by Zn2+ which should thus completely prevent o-PD photolabeling. In contrast, for b3-homomeric receptors, only three (or possibly four) of the five b3-H267 residues should be coordinated by Zn2+ (25) which should thus inhibit o-PD photolabeling by 60 to 80%. To test these predictions we photolabeled HEK cell membranes containing either a1b3 or b3-homomeric GABAA receptors with o-PD in the presence or absence of 100 mM Zn2+, a concentration that completely blocks current in both types of receptors (26, 27).
HEK cell membranes containing a1b3 GABAA receptors were photolabeled with 10 mM o-PD in the presence or absence of Zn2+ and the labeling efficiency of TM2-H267 was measured using middle-down mass spectrometry. TM2 was labeled with 0.63 ± 0.03% efficiency in the absence of Zn2+ and no photolabeling was detected in the presence of Zn2+ (Fig. 6A). To achieve a similar control photolabeling efficiency, membranes containing b3-homomeric GABAA receptors were photolabeled with 30 mM o-PD. The photolabeling efficiency of H267 was 0.95 ± 0.15%. In the presence of 100 mM ZnCl2, the photolabeling efficiency was decreased to 0.29 ± 0.03%, representing 70% inhibition of photolabeling. These results, along with the labeling stoichiometry determined by native MS, indicate that o-PD labels multiple H267 residues in a GABAA receptor pentamer. This is consistent with o-PD labeling from a pore-adjacent propofol binding site as opposed to a site within the pore.
Discussion
The goal of this study was to clarify the location and number
of specific propofol binding sites on GABAA receptors. The primary result is the validation and further characterization of
a specific propofol binding site adjacent to the 170 b3-H267 residue. The ability of propofol to protect H267 from photolabeling confirms that o-PD labeling identifies a specific propofol binding pocket (Fig. 2). This pocket appears to be ligand-specific, as the propofol analogue AziPM-d neither protects H267 from o-PD labeling nor labels nearby residues (Fig. 2). The photolabeling data are supported by molecular docking studies showing that propofol and o-PD assume energetically favorable poses in a pocket between the TM1, TM2, and TM3 of the b3 subunit in an a1b3 GABAA receptor, whereas AziPm does not (Fig. 4). This binding pocket is likely
- Figure 5. Native mass spectrometric analysis of photolabeling stoichiometry in homomeric b3 GABAA receptors. A, AziPm labeling: (Top panel) spectrum of unlabeled b3-ILC-del-homopentamer showing three charge-states (+27, +28, and +29). The calculated molecular mass of the pentamer is 215 kDa. (Lower panel) spectrum of b3-homopentamer photolabeled three times with 200 mM AziPm showing charge states +27, +28, and +29. Chargestate +28 shows 6 features, corresponding to the unlabeled receptor (dashed line) and five more features each with an additional mass of 216 ± 17 Da, consistent with AziPm adducts. B, same spectra as (A) focused on charge-state +28. The dashed line indicates the unlabeled b3-homopentamer and the numbers in red indicate the number of AziPm adducts in a b3-homopentamer.C, o-PD labeling: (Top Panel) spectrum of unlabeled b3-ICL-del homopentamer showing three charge-states (+29, +30, and +31) each corresponding to a molecular mass of 215 kDa. (Lower panel) spectrum of b3-ICL-del homopentamer photolabeled three times with 100 mM o-PD showing charge-states +29, +30, and + 31. Charge-state +30 shows four features, corresponding to the unlabeled receptor (dashed line) and three more features each with an additional mass of 232 ± 42 Da, consistent with o-PD adducts. D, same spectra as (C) focused on charge-state +30. The dashed line indicates the unlabeled b3-homopentamer and the numbers in red indicate the number of o-PD adducts in a b3-homopentamer. GABAA, gamma-aminobutyric acid type A; o-PD, ortho-propofol diazirine.
accessed via the pore, based on the channel-lining position of the o-PD photolabeled H267 residue and the absence of an apparent tunnel through which propofol could access this site from the membrane (22).
The ability of o-PD to label multiple H267 residues on a
GABAA receptor pentamer (at least 3 on a b3-homopentamer) is demonstrated by native MS (Fig. 5), suggesting that there
may be a pore-adjacent propofol binding site in each b3 subunit. Propofol must access these H267 residues from binding pockets surrounding the pore, since only a single propofol (or o-PD) molecule could be accommodated in the pore at the 170
position. Consistent with this idea, we found that when Zn2+ blocks the channel pore by forming a ternary complex with three H267 residues (24), the residual H267 residues in a b3-homopentamer can still be photolabeled by o-PD. This indicates that o-PD labeling of H267 must occur from sites surrounding the pore, since the pore itself is occluded by the Zn2+-H267 ternary complex and would not accommodate an o-PD molecule.
The proposition that o-PD labeling of H267 identifies specific propofol binding sites adjacent to the channel pore (9, 10) has been challenged with several arguments in addition to the
- Figure 6. The effect of ZnCl2 on o-PD photolabeling of b3-TM2 peptide in a1b3 b3-BRIL homomeric GABAA receptors. A, ZnCl2 completely inhibits oPD photolabeling of b3-TM2 peptide in a1b3 GABAA receptors. B, ZnCl2 partially ( 70%) inhibits o-PD photolabeling of b3-TM2 peptide in b3-homomeric GABAA receptors. a1b3 GABAA receptors were labeled with 10 mM o-PD and b3-homomeric receptors with 30 mM to achieve similar photolabeling efficiency. Samples with and without ZnCl2 (n = 3 for each) were compared using a unpaired t test with * = p < 0.05. GABAA, gamma-aminobutyric acid type A; o-PD, ortho-propofol diazirine; TM, transmembrane.
previously noted absence of propofol protection data. First, the finding that H267A (13) and H267C mutations (19) do not interfere with propofol enhancement of GABAA currents has been cited as evidence that H267 does not line a functionally important binding pocket. As observed in docking studies, the diazirine labeling group of o-PD is about 9 Å away from H267, suggesting it is not a crucial residue for propofol binding. Photolysis of o-PD generates a quinone-methide, a reactive intermediate that preferentially labels histidine and cysteine residues (28, 29). The quinone-methide is sufficiently longlived that it can diffuse to and label the nearby H267 residue; it must however be concentrated in a binding site proximal to H267, as o-PD does not label any of the numerous water-accessible histidine or cysteine residues on GABAA receptors. In contrast, b3 subunit residues Y143, F221, and Q224, which line the pore-adjacent binding pocket markedly reduce propofol enhancement of GABAA currents (13, 14, 16), supporting the proposed model. Second, the inability of propofol to protect H267C from modification by a water-soluble pCMBS regent has been interpreted as indicating that H267 labeling does not report on a propofol binding site (19). H267C lines the channel pore and should be readily modified by a water-soluble pCMBS regent in the pore; propofol in a poreadjacent binding pocket would not be expected to prevent this modification.
We also confirm the findings of a previous AziPm photolabeling study in which Edman degradation was used for sequencing (11). Our data show that AziPm and AziPM-d both
label the previously identified b3-M286, a1-I239, and b3-M227 residues, defining membrane-adjacent propofol binding sites
on the lipid face of a1b3 GABAA receptors in the b(+)/a(−), a(+)/b( ), and possibly b3(+)/b3( ) subunit interfaces. The mass spectrometric method provided complete sequence coverage of the TMDs of the a1 and b3 subunits and identified no additional labeled peptides. The additional mass units in AziPm-d (as compared to either AziPm or o-PD) facilitated identification of the labeled residues (Fig. 3C) and allowed us to examine the ability of o-PD to protect from AziPmd labeling. Both propofol and o-PD protected the M286, I239, and M227 residues from photolabeling, consistent with previous results obtained using radiolabeled AziPm (11). Docking studies showed that propofol, AziPm, and o-PD all bind in overlapping poses in the membrane-adjacent b(+)/a(−) and a(+)/b(−) propofol binding sites. Interestingly, the most efficiently labeled residue, I239, is two a-helical turns below the position of either the AziPm diazirine in its docked pose in an a1b3 GABAA receptor (Fig. 4C) or the propofol density in the cryogenic electron microscopy structure of an a1b2g2 GABAA receptor (17). This suggests that AziPm, may assume two poses in the b(+)/a(−) interface: one overlapping propofol in which it labels M286 and a second below propofol in the interface, in which it labels I239.
Collectively, the photolabeling and protection data indicate that o-PD binds to all of the identified propofol binding sites, whereas AziPM binds to the membrane-adjacent propofol binding sites, but not to the pore-adjacent site. These data
highlight the limitations of both photolabeling reagents. o-PD closely mimics the positive allosteric effects of propofol on GABAA currents (Table 1), but due to photochemical specificity, labels only a subset of the sites to which it binds. In contrast, AziPm is an effective photolabeling reagent that can label any carbon-hydrogen bond (and hence can label any amino acid) but binds to only a subset of propofol binding sites and weakly mimics the positive allosteric effects of propofol (Table 1). AziPm is a weak positive allosteric modulator of GABA response in a1b3 GABAA receptors (Table 1) (12) with lower potency and efficacy than either propofol or o-PD. Moreover, it produces virtually no direct activation of the channels, even at 100 mM. The absence of binding to the poreadjacent site offers one potential explanation for the low efficacy of AziPm. Alternatively, the low efficacy of AziPm could be a consequence of low efficacy in one or both of the membrane-adjacent sites. It is difficult to distinguish between these two mechanisms, and both may contribute to the low efficacy of AziPm.
How many sites contribute to the positive allosteric modulator effects of propofol? The number of sites to which propofol binds on a GABAA receptor depends on the subunit composition. A b3 homomeric receptor has five pore-adjacent
- b3 subunit sites and five membrane-adjacent b3(+)/b3(−) sites for a total of ten sites per pentamer; an a1b3 receptor has five membrane-adjacent sites [a mixture of b3(+)/a1(−), a1(+)/ b3(−) and b3(+)/b3(−)] and three pore-adjacent b-subunit sites for a total of eight sites per pentamer; and an a1b3g2 receptor has two b3(+)/a1(−), one a1(+)/b3(−) and possibly one g2(+) b3(−) membrane-adjacent sites, as well as two pore-adjacent b3-subunit sites for a total of five or six propofol binding sites per pentamer.
Which of these binding sites contribute to the positive
allosteric effect of propofol? b3-homomeric receptors provide the simplest case to study with only two types of sites: pore-
adjacent and b3(+)/b3(−) membrane-adjacent sites. Tryptophan mutations of the Y143, F221, and Q224 residues that line the pore-adjacent site (Fig. 4, A and B) all markedly reduce propofol, but not pentobarbital activation of currents (13). The Y143W and Q224W mutations preferentially reduce activation by bulky propofol analogues, suggesting steric interference with propofol binding by the tryptophan mutations and indicating a functional contribution of the pore-adjacent sites to propofol action. In a1b3 GABAA receptors, individual amino acid substitutions in either the pore-adjacent site (Q224W, Y143W, and F221W) or the b3(+)/a1(−) membrane-adjacent site (M286W) have minimal effect on propofol activation, whereas any combination of mutations in the two sites (e.g. Y143W/M286W) virtually eliminates propofol activation (14). These data indicate that both the b3(+)/a1(−) membraneadjacent sites and the pore-adjacent sites contribute to propofol activation. Finally, a study in a1b2g2 receptors used concatemeric b2-a1-g2 and b2-a1 constructs with mutations of b2-Y143W and/or b2-M286W to probe the number of functional propofol binding sites and their energetic contributions to propofol gating (16). The results indicate that the two pore adjacent sites and the two b2(+)/a1(−) membrane adjacent
sites contribute equally and additively to channel activation. Analysis of the data in a Monod–Wyman–Changeux framework was best fit by a model with five or six (rather than four) functionally equivalent propofol sites. The a1(+)/b2(−) membrane-adjacent site is a logical candidate for the additional functional binding site(s) predicted by this model. While a b3-M227W mutation has been reported to have no qualitative effect on propofol activation of a1b3g2 receptors (30), the effect of mutations in this site have not been fully evaluated.
A cryogenic electron microscopic structure of a desensitized a1b2g2 GABAA receptor with a propofol density in the b2(+)/ a1(−) membrane-accessible site was recently published (17). Propofol densities were detected in neither the pore-adjacent site nor the a1(+)/b2(−) membrane-adjacent site. Why was propofol not observed in these sites that have been identified by photolabeling (9, 11) and supported by molecular docking (10) and mutational (13, 14, 16) studies? It should be appreciated that when a receptor is photolabeled in a membrane or detergent, the photolabeling reagent can sample multiple conformations of the receptor. In contrast, structural analyses usually identify a single protein conformation, which may or may not be the preferred conformation for ligand binding. Notably, while several structures of GABAA receptors have been published (17, 31–38), there has not been a structure of a nondesensitized open receptor. It is plausible that propofol preferentially binds to the pore-adjacent and a1(+)/b3(−) membrane accessible sites in the open state relative to the desensitized state. Indeed, propofol decreases the rate and extent of GABAA receptor desensitization indicating that it should have state-dependent binding that favors the open state (39, 40). The binding of estradiol to a pore-adjacent binding site on the GABA-r1 receptor illustrates the conformationdependent binding of an allosteric modulator to a pentameric ligand-gated ion channel. Estradiol, an inhibitor of r1 currents, is observed in the cryo-EM structures of the resting and preopen conformations of the r1, but not in the (post open) desensitized conformation (41) A second reason why the propofol sites identified by photolabeling may not be observed in cryo-Em structures is that propofol is a small molecule with low (mM) affinity for its binding sites and may exhibit multiple binding poses. The b2(+)/a1(−) membrane-adjacent site has been estimated to have a 2-fold higher affinity for propofol than the pore-adjacent site (16), possibly making it easier to detect. Hopefully, as structural resolution continues to improve, the molecular details and state-dependence of propofol binding in each of its sites will become available.
Experimental procedures
Synthesis of o-PD o-PD was synthesized as previously described (9).
Synthesis of AziPm and AziPm-d9
The synthesis of AziPm was carried out according to the literature method (12). The nona-deuterated version (AziPmd9, I) was synthesized similarly from cumene-d11 (IV). This was made, in turn, from benzene-d6 (Fig. 7) by way of
sequential bromination, Grignard formation, and reaction with acetone-d6, followed by reduction with triphenylphosphine/ iodine (42).
All reagents and solvents were used as bought from commercial sources. 1H, 13C, and 19F NMR spectra were recorded on a JEOL 400 MHz nuclear magnetic resonance spectrometer. Mass spectra were recorded in either positive or negative mode by electron ionization or electron spray ionization methods. AziPm HRMS negative ion spectroscopy showed m/ z = 243.0753; m/z calculated for [C11H11F3N2O-H] = 243.0751.
Bromobenzene-d5
Benzene-d6 (25.0 g, 0.297 mol) and pyridine (250 mg 3.16 mmole) were added to a three-necked round-bottomed flask fitted with a magnetic stirrer bar. Bromine (62.4 g, 0.39 mol) was added slowly with delayed evolution of a white gas. After addition of the bromine, the solution was stirred at 40 C for 30 min followed by 1 h at 70 C. The contents of the flask were washed with three aliquots of dilute sodium hydroxide solution followed by washing once with water. The organic layer was dried over anhydrous calcium chloride. The liquid was then distilled under water-pump vacuum to give a fraction boiling at 30 to 40 C. Further distillation under atmospheric pressure gave a fraction boiling at 154 to 158 C (21.77 g, 45% yield). Electron-ionization MS of the liquid showed two peaks m/z = 160.15 and 162.15 m/z (Calc. for C6D5Br (M + H) = 160.9949-, 162.9618).
2-Phenyl-2-propanol-d11
Bromobenzene-d5 (20.9 g; 0.129 mol) was dissolved in dry diethyl ether (10 ml) in a dropping funnel connected to a twonecked round bottom flask equipped with a magnetic stirrer bar and a water-cooled condenser. The flask was charged with magnesium filings (3.8 g; 0.155 mol) and dry diethyl ether (20 ml). A few drops of the bromobenzene-d5 solution were added to the mixture and immediate reflux of the diethyl ether was noted. Addition of the remaining bromobenzene-d5 was continued at a rate to keep the diethyl ether boiling vigorously. On completion of the addition, the brown solution was stirred for 15 min. Acetone-d6 was added at a rate to restart and maintain the boiling of the diethyl ether. The solution was stirred for 2 hours and then poured into 2M hydrochloric acid (75 ml). The organic layer was separated. The aqueous phase was extracted twice with diethyl ether, the organic layers
combined and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure to give 20 g of an orange liquid which was used without further purification in the next step of the synthesis.
Cumene-d11
The product from the previous step (20 g; 136 mmol) was added with triphenylphosphine (42.9 g; 151.6 mmol), iodine (41.6 g; 163.8 mmol), and benzene (350 ml) into a roundbottom flask equipped with a stirrer bar and water-cooled condenser. The mixture was refluxed for 150 min. After cooling, the mixture was shaken twice with a solution of sodium sulfite to remove iodine. The yellow organic layer was separated and washed once with water. Benzene was removed under reduced pressure to give an orange solid. This solid was then heated under reduced pressure to give two fractions of a light-yellow liquid, one boiling at 56 to 60 C (F1) and the second boiling at 64 to 84 C (F2). The two fractions were subjected to proton NMR. F2 showed a peak at 2.9 ppm which is consistent with the desired cumene-d11 (2.53 g, 16% yield).
p-Trifluoroacetylcumene-d11 and AziPm-d
An equivalent molar quantity of undeuterated cumene was added to the crude cumene-d11 (2.53 g; 19 mmol), thereby allowing a more definitive identification of the mass spectral signal of the AziPm when bound to any given peptide from the GABAA receptor. The mixture was dissolved in dichloromethane (60 ml) and 4-(dimethylamino)pyridine (4.9 g; 40 mmol) was added, followed by trifluoroacetic anhydride (8.76 g; 42 mmol). The solution was cooled to 0 C in an ice bath followed by addition of anhydrous aluminum chloride (15.9 g; 119 mmol) when the solution slowly turned dark brown. The solution was cooled further to −10 C in an acetone/ice bath for 15 min after which it was allowed to warm to room temperature and stirred for 5 days. The solution was poured into ice/water (500 ml) and the organic layer separated. The aqueous layer was extracted with two aliquots of dichloromethane, and the organic layers combined and dried over anhydrous sodium sulfate. This was then used without further purification for the continued synthesis of I. A negative ion, high resolution mass spectrum of the AziPm-d product showed features at m/z values of 250.1185, 251.1250, 252.1320, and 252.1380 with intensities in a ratio of 21:40:27:12. The calculated m/z for AziPm-d9 [C11H2D9F3N2O -H] = 252.1316. These data
indicate that the final product is a mixture of AziPm-d6, d7, d8, and d9. with AziPm-d7 and AziPm-d8 being the most abundant species. The reagent is hence referred to as AziPm-d.
Cell culture, protein expression and membrane preparation
Tetracycline inducible HEK-T-Rex-293 cells stably expressing high-density human a1-8xHis-FLAG and human b3 GABAA receptor subunits were established as reported previously (20). Briefly, the cells were cultured in Dulbecco’s modified Eagle’s medium/F-12 50/50 medium containing 10% fetal bovine serum (tetracycline-free, Takara), penicillin (100 units/ml), streptomycin (100 g/ml), blasticidin (2 mg/ml), hygromycin (50 mg/ml), and zeocin (20 mg/ml) at 37 C in a humidified atmosphere containing 5% CO2. Upon reaching 50% confluence, GABAA receptor expression was induced by treating the cells with 1 mg/ml doxycycline with the addition of 5 mM sodium butyrate for 48 to 72 h. After induction, the cells were harvested in a buffer containing 10 mM potassium phosphate, 100 mM potassium chloride (pH 7.5) plus protease inhibitors (Sigma-Aldrich). The cells were collected by centrifugation at 1000g at 4 C for 5 min and homogenized with a glass mortar and a Teflon pestle for ten strokes on ice. Membranes were collected by centrifugation at 40,000g at 4 C for 30 min and resuspended in a buffer containing 10 mM potassium phosphate and 100 mM potassium chloride (pH
- 7.5). Protein concentration was determined with a microbicinchoninic acid protein assay (Thermo Fisher Scientific). GABAA receptor density was determined by measuring the Bmax of [3H]muscimol binding as previously described (20). Membranes proteins were stored at −80 C.
Two human b3 constructs were designed to be expressed using baculovirus in HEK GnTI- cells. The intracellular loop was deleted between residues F307 and A421of the mature peptide and replaced with a short sequence, SQPARAA. Additionally, the 1D4 rhodopsin epitope sequence was added to the complementary DNA (cDNA) to produce a protein with the epitope added to the C terminus for protein purification. This construct has been named hb3-ICL-del 1D4. Another cDNA construct included the BRIL sequence in the intracellular loop region between residues A325 and A421 for the purpose of stabilizing the receptor and is referred to as b3BRIL (24). Both of these genes were synthesized by Twist Bioscience using the pEG BacMam vector designed by Eric Gouaux (43).
Bacmid viral DNA was produced by transforming Escherichia coli containing the DH10EMBacVSV bacmid with the cDNA. Using the beta galactosidase blue/white colony selection in the presence of gentamycin, tetracycline, and kanamycin, white colonies were selected that indicated insertion of the cDNA into the transposon elements of the bacmid. These were selected and cultured in LB medium. DNA was prepared from harvested cells, and PCR was done as a quality control to ensure proper insertion and that a full-length gene was present. Bacmid was transfected into Sf9 cells to make the first generation of viral particles (P0). This initial viral culture was introduced into a suspension culture of Sf9 cells (150 ml) with a density of 1 ×
106 cells/ml and multiplicity of infection of 0.005. After approximately 6 days, a red color was observed from the coproduction of mcherry fluorescent protein. The viral particles were purified from the Sf9 cells and used to transduce expression in HEK GnTI- cells in suspension at a density of 2 × 106 cells/ml with a multiplicity of infection of 2 on day one. Cells were harvested on day 5, approximately 90 h after induction, by centrifugation at 1000g. After removing the supernatant, cells were washed with PBS and resuspended in PBS with protease inhibitors, centrifuged again, and frozen in liquid nitrogen.
Photolabeling and purification of a1b3 and b3-homomeric GABAA receptors for middle-down MS analysis
For photolabeling experiments, 20 to 40 mg of HEK cell membrane proteins (about 150 pmol of receptor as determined by [3H]muscimol binding) were thawed and resuspended in a buffer (10 mM potassium phosphate, 100 mM potassium chloride (pH 7.5), and 1 mM GABA) at a final concentration of 1.25 mg/ml. The membranes were incubated with the photolabeling analogues on ice for 1 h. For experiments examining competitive prevention of photolabeling, membranes were incubated at 4◦C for 1 h with o-PD or AziPm-d in the presence or absence of a putative competitor (300 mM propofol, o-PD and AziPm-d9, or 100 mM ZnCl2); an equal volume of vehicle (ethanol) was added to all samples. The samples were then irradiated in a quartz cuvette for 5 min, using a photoreactor emitting light at >320 nm as previously described (44). The membranes were then collected by centrifugation at 20,000g for 45 min at 4 C. Membrane proteins were solubilized in lysis buffer containing 1% DDM, 0.25% cholesteryl hemisuccinate, 50 mM Tris (pH 7.5), 150 mM NaCl, 2 mM CaCl2, 5 mM KCl, 5 mM MgCl2, 1 mM EDTA, and 10% glycerol at a final concentration of 1 mg/ml. The membrane suspension was homogenized using a glass mortar and a Teflon pestle and incubated at 4 C overnight. The protein lysate was centrifuged at 20,000g for 45 min at 4 C. The resulting supernatant was incubated for 2 h at 4 C with 0.2 ml anti-FLAG agarose (Sigma-Aldrich). The anti-FLAG agarose was then transferred to an empty column and washed with 20 ml of washing buffer (50 mM triethylammonium bicarbonate and 0.05% DDM). GABAA receptors were eluted with aliquots of washing buffer containing 200 mg/ml FLAG tag peptide and 100 mg/ml 3X FLAG (ApexBio). The pooled eluates (4 ml) containing GABAA receptors were concentrated to 100 ml using a 100 kDa cut-off centrifugal filter. For homomeric b3-GABAA receptor purification, membrane proteins were solubilized in the same lysis buffer used with a1b3-GABAA receptors and the protein lysate was incubated with Rho-1D4 agarose at 4 C for 6 h. After washing with 20 ml washing buffer, b3-GABAA receptors were eluted with washing buffer containing 500 mM Rho-1D4 peptide at 4 C overnight.
Middle-down MS analysis
A middle-down mass spectrometric method was used to selectively detect and sequence the TMDs of GABAA receptors as previously described (20). Affinity-enriched GABAA
receptors were reduced with 5 mM tris(2-carboxyethyl)phosphine for 1 h, alkylated with 5 mM NEM for 1 h in the dark, and quenched with 5 mM DTT for 15 min. These three steps were done at room temperature. Samples were then digested with 8 mg of trypsin for 7 days at 4 C. The digestions were terminated by adding formic acid in a final concentration of 1%, followed directly by LC-MS analysis on an Orbitrap Elite mass spectrometer. Subsequently, 20 ml samples were injected onto a home-packed PLRP-S (Agilent Technologies) column (10 cm × 75 mm, 300 Å), separated with a 145 min gradient from 10% to 90% acetonitrile, and introduced to the mass spectrometer at 800 nl/min with a nanospray source. The survey MS1 scans were acquired at high resolution (60,000 resolution) in the range of m/z = 100 to 2000 and the fragmentation spectra were acquired at 15,000 resolution. Datadependent acquisition of the top 20 MS1 precursors with exclusion of singly charged precursors was set for MS2 scans. Fragmentation was performed using collision-induced dissociation or high-energy dissociation with normalized energy of 35%. The data were acquired and reviewed with Xcalibur 2.2 (Thermo Fisher Scientific).
The LC-MS data were searched against a customized database containing the sequence of the GABAA receptor a1-8XHis-FLAG and b3 subunits and filtered with 1% false discovery rate using PEAKS Xpro (Bioinformatics Solutions Inc). Search parameters were set for a precursor mass accuracy of 20 ppm, fragmentation ion accuracy of 0.1 Da, up to three missed cleavages on either side of peptides with trypsin digestion. Methionine oxidation, cysteine alkylation with NEM or DTT, and adducts of o-PD (mass = 216.08), AziPm (mass = 216.08), AziPm-d7 (mass = 223.13), or AziPm-d8 (mass = 224.13) on any amino acid were included as variable modifications. Photolabeling efficiency was estimated by measuring the areas under the curve (AUC) of selected ion chromatograms of the photolabeled and corresponding unlabeled peptides and calculating the ratio of the labeled peptide AUC/(labeled peptide AUC + unlabeled peptide AUC). In AziPm-d labeling experiments, photolabeling efficiency was estimated using the sum of the AUC for AziPm-d7 and AziPmd8 adducts. Results are expressed as percent efficiency. Efficiency of photolabeling between conditions was compared using paired t test for binary comparisons (Prism 10, GraphPad Software, San Diego, CA; https://www.graphpad.com/ scientific-software/prism/). The MS2 spectra of photolabeled TMD peptides were also manually analyzed for fragment ion charge state and mass accuracy and to confirm the sequence assignment and sites of adduction.
Native MS of photolabeled b3-homomeric GABAA receptors
For native MS experiments, b3-ICL-del receptors were solubilized in 1% 2,2-dioctylpropane-1,3-bis-b-D-maltopyranoside (DMNG), affinity-purified on Rho-1D4 antibodycoupled beads followed by further purification by sizeexclusion chromatography (22). Subsequently, 30 mg of purified human b3-ICL-del GABAA receptor in 75 ml buffer (10 mM Hepes, pH 7.2, 150 mM NaCl, 0.007% (w/v) DMNG,
0.0006% (w/v) cholesteryl hemisuccinate) was preincubated with photolabeling reagent at 4 C for 30 min prior to UVirradiation. To achieve maximum photolabeling efficiency, AziPm was added to the protein at a concentration of 200 mM three times with UV-irradiation between each application. o-PD was applied to the sample at a concentration of 100 mM three times with UV-irradiation between each application. A lower concentration of o-PD was necessary to perform native MS analysis. Even at this concentration, the native MS spectrum for the o-PD sample showed more noise, possibly due to a detrimental effect of free o-PD on the electrospray ionization process. The AziPm and o-PD photolabeled samples were prepared from two separate b3 GABAA receptor purifications, and nonphotolabeled b3 GABAA receptor samples were analyzed in each case. After photolabeling, the samples were buffer exchanged into 200 mM ammonium acetate pH 8 and 0.08 mM DMNG ( 2 times critical micelle concentration [CMC]) using Bio-Spin gel filtration columns (Bio-Rad). Briefly, 3 ml of this sample was directly loaded into a borosilicate capillary emitter (Thermo Fisher Scientific, ES380) and native MS spectra were obtained by static nanospray on a Thermo Q-Exactive EMR mass spectrometer. The data were collected using an electrospray voltage of 1.2 to 1.5 kV, capillary temperature of 200 C, resolution of 8750, trap and transfer voltages of 200 V (CID) and 150 V (CE), respectively, for the Azi-Pm sample. For the o-PD sample, the transfer voltage was increased to 200 V. Ion transfer optics were set at 8, 7, 6, and 4 V for injection flatapole, interflatapole lens, bent flatapole, and transfer multiple, respectively. Since the o-PD spectrum had significant noise, all spectra were deconvoluted manually using the three observed charge states, and the molecular weight determined by averaging the mass from the three charge states. For the peak corresponding to 2 o-PD labels in the b3 GABAA receptor pentamer, only the +30and +31-charge states were used, and for the peak corresponding to 3 o-PD labels, only the +30-charge state was used. The observed charge states differed between the experiments performed with o-PD and Azi-Pm. This variability is commonly observed and may be a consequence of differences in the protein sample between preparations or variations in the capillary emitter tip structure.
Docking simulations
The molecular coordinates of propofol, o-PD, and AziPm were generated by Avogadro software (https://avogadro.cc/) with minimized energy and converted into a PDB file. A model of the human a1b3 GABAA receptor in the presence of GABA (Protein Data Base bank ID: 7PBD) was used for docking. Docking preparation including deletion of water and addition of charges was performed in Autodock tools. All three ligands as well as amino acid residues of GABAA receptors surrounding the photolabeling sites were set as flexible in Autodock tools. The flexible residues for the pore-adjacent site are as follows: b3Y143, b3Y220, b3Q224, b3Th225, b3P228, b3T263, b3I264, b3H267, b3L268, b3I281, a1T267, a1I271, and a1N275t; for the membrane-adjacent a1(+)/b3(−) site: b3I222,
b3L223, b3Q224, b3M227, b3P228, b3I230, b3L231, a1D287, a1W288, a1A291, a1V292, a1Y294, and a1A295; and for the membrane-adjacent b3(+)/a+(−) site: b3M283, b3M286, b3V290, a1I228, a1L232, a1l235, a1M236, and a1I239. The docking was performed using AutoDock Vina (https://vina. scripps.edu/) (45). Docking grid boxes were built to cover the whole TMD domain with dimensions of 50 × 50 × 45 Å for initial search and 25 × 25 × 25 Å for each individual site encompassing all the flexible amino acid residues. Docking was limited to an energy range of 4 kcal from the best docking pose. Docking poses were visualized in UCSF chimera 1.16 and ChimeraX.
Receptor expression in Xenopus laevis oocytes and electrophysiological recordings
Human a1b3 GABAA receptors were expressed in oocytes from the African clawed frog (X. laevis). Oocyte preparation and complementary RNA injections were done as described in detail previously (46). The complementary RNA injection ratio was 5:1 (a1:b3, 2.5 ng: 0.5 ng) to reduce the expression of homomeric b3 receptors. The electrophysiological recordings were conducted using standard two-electrode voltage clamp. The oocytes were clamped at −60 mV and perfused with ND96 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM Hepes; pH 7.4) at 5 to 8 ml/min. Solutions were gravityapplied from glass syringes with glass luer slips via Teflon tubing. A typical potentiation experiment consisted of exposing an oocyte to 0.2 to 0.3 mM GABA (EC7-9), followed by coapplications of GABA and test compound and, for normalization purposes, an application of 30 mM (saturating) GABA. Direct activation experiments consisted of application of 100 mM test compound followed by application of 30 mM GABA. Drug applications were separated by 2 to 3 min washes in ND96. The current responses were amplified with an OC-725C amplifier (Warner Instruments), digitized with a Digidata 1200 series digitizer (Molecular Devices), and stored using pCLAMP (Molecular Devices; https://www. moleculardevices.com/products/axon-patch-clamp-system/ acquisition-and-analysis-software/pclamp-software-suite). The analysis of current traces was done with Clampfit (Molecular Devices). The stock solution of GABA was made in ND96 bath solution at 500 mM, stored in aliquots at −20 C, and diluted as needed on the day of experiment. The steroids were dissolved in dimethyl sulfoxide at 10 to 20 mM and stored at room temperature.
Data availability
All the data described in this manuscript is contained within the manuscript. Raw mass spectrometry data files will be shared upon request to the corresponding author (eversa@ wustl.edu).
Acknowledgments—We express our appreciation to Joe Henry Steinbach for his thoughtful comments and suggestions on the manuscript and to Lei Wang for experimental assistance.
Author contributions—Z.-W. C., S. M. C., J. B., Y. S., S. R. P., B. R. V., W. W. L. C., and A. S. E. investigation; Z.-W. C., S. M. C., J. B., Y. S., S. R. P., B. R. V., W. W. L. C., and G. A. methodology; Z.-W. C., N. P. F., W. W. L. C., and G. A. writing–review and editing; Z.-W. C., W. W. L. C., and G. A. formal analysis; Z.-W. C. and A. S. E. conceptualization; Z.-W. C. and A. S. E. writing–original draft; Z.-W. C. and A. S. E. visualization; Z.-W. C. validation; J. B., B. R. V., E. H. S., C. J. E., and N. P. F. resources; N. P. F., G. A., and A. S. E. funding acquisition; N. P. F., G. A., and A. S. E. supervision; A. S. E. project administration.
Funding and additional information—This work was funded by NIH RO1GM108799 (A. S. E.), R35GM149287 (A. S. E.), R35GM140947 (G. A.), Wellcome Trust 220759/Z/20/Z (N. P. F.), and the Taylor Institute for Innovative Psychiatric Research. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
Abbreviations—The abbreviations used are: AUC, areas under the curve; cDNa, complementary DNA; DDM, n-dodecyl-beta-maltoside; DMNG, 2,2-dioctylpropane-1,3-bis-b-D-maltopyranoside; GABAA, gamma-aminobutyric acid type A; MS, mass spectrometry; NEM, N-ethylmaleimide; o-PD, ortho-propofol diazirine; TMD, transmembrane domain.
References
- 1. Krasowski, M. D., Jenkins, A., Flood, P., Kung, A. Y., Hopfinger, A. J., and Harrison, N. L. (2001) General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility. J. Pharmacol. Exp. Ther. 297, 338 351
- 2. Jurd, R., Arras, M., Lambert, S., Drexler, B., Siegwart, R., Crestani, F., et al. (2003) General anesthetic actions in vivo strongly attenuated by a point mutation in the GABA(A) receptor beta3 subunit. FASEB J. 17, 250–252
- 3. Hales, T. G., and Lambert, J. J. (1991) Modulation of the gabaareceptor by propofol. Anesthesiology 75, A587
- 4. Sanna, E., Mascia, M. P., Klein, R. L., Whiting, P. J., Biggio, G., and Harris, R. A. (1995) Actions of the general anesthetic propofol on recombinant human GABAA receptors: influence of receptor subunits. J. Pharmacol. Exp. Ther. 274, 353–360
- 5. Ruesch, D., Neumann, E., Wulf, H., and Forman, S. A. (2012) An allosteric coagonist model for propofol effects on a1b2g2L g-aminobutyric acid type A receptors. Anesthesiology 116, 47 55
- 6. Chua, H. C., and Chebib, M. (2017) GABAA receptors and the diversity in their structure and pharmacology. Adv. Pharmacol. 79, 1 34
- 7. Nys, M., Kesters, D., and Ulens, C. (2013) Structural insights into Cysloop receptor function and ligand recognition. Biochem. Pharmacol. 86, 1042–1053
- 8. Scott, S., and Aricescu, A. R. (2019) A structural perspective on GABAA receptor pharmacology. Curr. Opin. Struct. Biol. 54, 189–197
- 9. Yip, G. M. S., Chen, Z.-W., Edge, C. J., Smith, E. H., Dickinson, R., Hohenester, E., et al. (2013) A propofol binding site on mammalian GABAA receptors identified by photolabeling. Nat. Chem. Biol. 9, 715–720
- 10. Franks, N. P. (2015) Structural comparisons of ligand-gated ion channels in open, closed, and desensitized states identify a novel propofol-binding site on mammalian g-aminobutyric acid type A receptors. Anesthesiology 122, 787 794
- 11. Jayakar, S. S., Zhou, X., Chiara, D. C., Dostalova, Z., Savechenkov, P. Y., Bruzik, K. S., et al. (2014) Multiple propofol-binding sites in a
- g-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog. J. Biol. Chem. 289, 27456–27468
- 12. Hall, M. A., Xi, J., Lor, C., Dai, S., Pearce, R., Dailey, W. P., et al. (2010) mAzipropofol (AziPm) a photoactive analogue of the intravenous general anesthetic propofol. J. Med. Chem. 53, 5667–5675
- 13. Eaton, M. M., Cao, L. Q., Chen, Z., Franks, N. P., Evers, A. S., and Akk, G.
- 14. Eaton, M. M., Germann, A. L., Arora, R., Cao, L. Q., Gao, X., Shin, D. J., et al. (2016) Multiple non-equivalent interfaces mediate direct activation of GABAA receptors by propofol. Curr. Neuropharmacol. 14, 772–780
- 15. Krasowski, M. D., Nishikawa, K., Nikolaeva, N., Lin, A., and Harrison, N. L. (2001) Methionine 286 in transmembrane domain 3 of the GABAA receptor b subunit controls a binding cavity for propofol and other alkylphenol general anesthetics. Neuropharmacology 41, 952–964
- 16. Shin, D. J., Germann, A. L., Johnson, A. D., Forman, S. A., Steinbach, J. H., and Akk, G. (2018) Propofol is an allosteric agonist with multiple binding sites on concatemeric ternary GABAA receptors. Mol. Pharmacol. 93, 178 189
- 17. Kim, J. J., Gharpure, A., Teng, J., Zhuang, Y., Howard, R. J., Zhu, S., et al.
- 18. Tang, P., and Eckenhoff, R. (2018) Recent progress on the molecular pharmacology of propofol. F1000Res 7, 123
- 19. Stern, A. T., and Forman, S. A. (2016) A cysteine substitution probes b3H267 interactions with propofol and other potent anesthetics in a1b3g2L g-aminobutyric acid type A receptors. Anesthesiology 124, 89–100
- 20. Chen, Z.-W., Bracamontes, J. R., Budelier, M. M., Germann, A. L., Shin, D. J., Kathiresan, K., et al. (2019) Multiple functional neurosteroid binding sites on GABAA receptors. PLoS Biol. 17, e3000157
- 21. Chen, Z.-W., Fuchs, K., Sieghart, W., Townsend, R. R., and Evers, A. S.
- 22. Miller, P. S., and Aricescu, A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275
- 23. Reeves, P. J., Callewaert, N., Contreras, R., and Khorana, H. G. (2002) Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracyclineinducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. U. S. A. 99, 13419–13424
- 24. Kasaragod, V. B., Mortensen, M., Hardwick, S. W., Wahid, A. A., Dorovykh, V., Chirgadze, D. Y., et al. (2022) Mechanisms of inhibition and activation of extrasynaptic ab GABAA receptors. Nature 602, 529–533
- 25. Vallee, B. L., and Auld, D. S. (1990) Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry 29, 5647 5659
- 26. Hosie, A. M., Dunne, E. L., Harvey, R. J., and Smart, T. G. (2003) Zincmediated inhibition of GABA(A) receptors: discrete binding sites underlie subtype specificity. Nat. Neurosci. 6, 362–369
- 27. Wooltorton, J. R., Moss, S. J., and Smart, T. G. (1997) Pharmacological and physiological characterization of murine homomeric beta3 GABA(A) receptors. Eur. J. Neurosci. 9, 2225–2235
- 28. Bolton, J. L., Turnipseed, S. B., and Thompson, J. A. (1997) Influence of quinone methide reactivity on the alkylation of thiol and amino groups in proteins: studies utilizing amino acid and peptide models. Chem. Biol. Interact. 107, 185–200
- 29. Modica, E., Zanaletti, R., Freccero, M., and Mella, M. (2001) Alkylation of amino acids and glutathione in water by o-quinone methide. Reactivity and selectivity. J. Org. Chem. 66, 41–52
- 30. Nourmahnad, A., Stern, A. T., Hotta, M., Stewart, D. S., Ziemba, A. M., Szabo, A., et al. (2016) Tryptophan and cysteine mutations in M1 helices of a1b3g2L g-aminobutyric acid type A receptors indicate distinct intersubunit sites for four intravenous anesthetics and one orphan site. Anesthesiology 125, 1144 1158
- 31. Zhu, S., Noviello, C. M., Teng, J., Walsh, R. M., Jr., Kim, J. J., and Hibbs, R. E. (2018) Structure of a human synaptic GABAA receptor. Nature 559, 67–72
- 32. Zhu, S., Sridhar, A., Teng, J., Howard, R. J., Lindahl, E., and Hibbs, R. E.
- 33. Legesse, D. H., Fan, C., Teng, J., Zhuang, Y., Howard, R. J., Noviello, C. M., et al. (2023) Structural insights into opposing actions of neurosteroids on GABAA receptors. Nat. Commun. 14, 5091
- 34. Phulera, S., Zhu, H., Yu, J., Claxton, D. P., Yoder, N., Yoshioka, C., et al.
- 35. Sun, C., Zhu, H., Clark, S., and Gouaux, E. (2023) Cryo-EM structures reveal native GABAA receptor assemblies and pharmacology. Nature 622, 195–201
- 36. Laverty, D., Desai, R., Uchanski, T., Masiulis, S., Stec, W. J., Malinauskas, T., et al. (2019) Cryo-EM structure of the human a1b3g2 GABAA receptor in a lipid bilayer. Nature 565, 516–520
- 37. Masiulis, S., Desai, R., Uchanski, T., Serna Martin, I., Laverty, D., Karia, D., et al. (2019) GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 565, 454–459
- 38. Sente, A., Desai, R., Naydenova, K., Malinauskas, T., Jounaidi, Y., Miehling, J., et al. (2022) Differential assembly diversifies GABAA receptor structures and signalling. Nature 604, 190–194
- 39. Bai, D., Pennefather, P. S., MacDonald, J. F., and Orser, B. A. (1999) The general anesthetic propofol slows deactivation and desensitization of GABAAReceptors. J. Neurosci. 19, 10635 10646
- 40. Orser, B. A., Wang, L. Y., Pennefather, P. S., and MacDonald, J. F. (1994) Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. J. Neurosci. 14, 7747–7760
- 41. Fan, C., Cowgill, J, Howard, R. J., and Lindahl, E. (2024) Divergent mechanisms of steroid inhibition of human r1 receptors. Nat. Commun. 15, 5216–5229
- 42. Furukawa, I., Zhou, B., and Hashimoto, S. (1984) Reaction of alcohols with triphenylphosphine diiodide. Nippon Kagaku kaishi 7, 1208–1212
- 43. Goehring, A., Lee, C.-H., Wang, K. H., Michel, J. C., Claxton, D. P., Baconguis, I., et al. (2014) Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat. Protoc. 9, 2574–2585
- 44. Germann, A. L., Pierce, S. R., Tateiwa, H., Sugasawa, Y., Reichert, D. E., Evers, A. S., et al. (2021) Intrasubunit and intersubunit steroid binding sites independently and additively mediate a1b2g2L GABAA receptor potentiation by the endogenous neurosteroid allopregnanolone. Mol. Pharmacol. 100, 19–31
- 45. Trott, O., and Olson, A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461
- 46. Pierce, S. R., Germann, A. L., Steinbach, J. H., and Akk, G. (2022) The sulfated steroids pregnenolone sulfate and dehydroepiandrosterone sulfate inhibit the a1b3g2L GABAA receptor by stabilizing a novel nonconducting state. Mol. Pharmacol. 101, 68–77
Figures
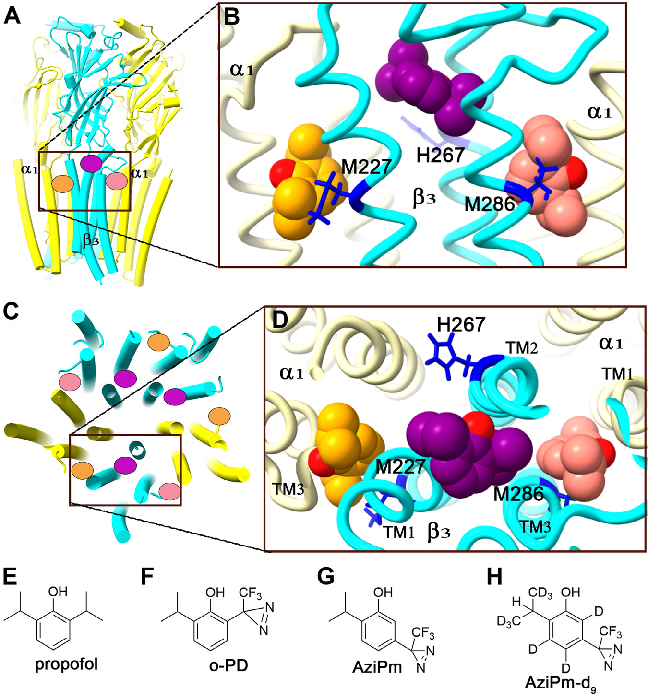
Figure 4
Electrophysiology or binding affinity data for propofol at different GABAA receptor binding sites, comparing potency across the three identified site classes.
chart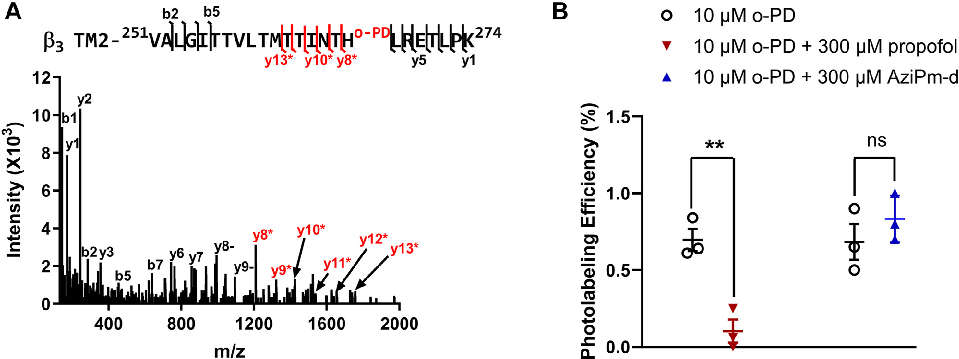
Figure 5
Mutagenesis or competition binding data supporting the classification of three distinct propofol binding site classes on GABAA receptors.
chart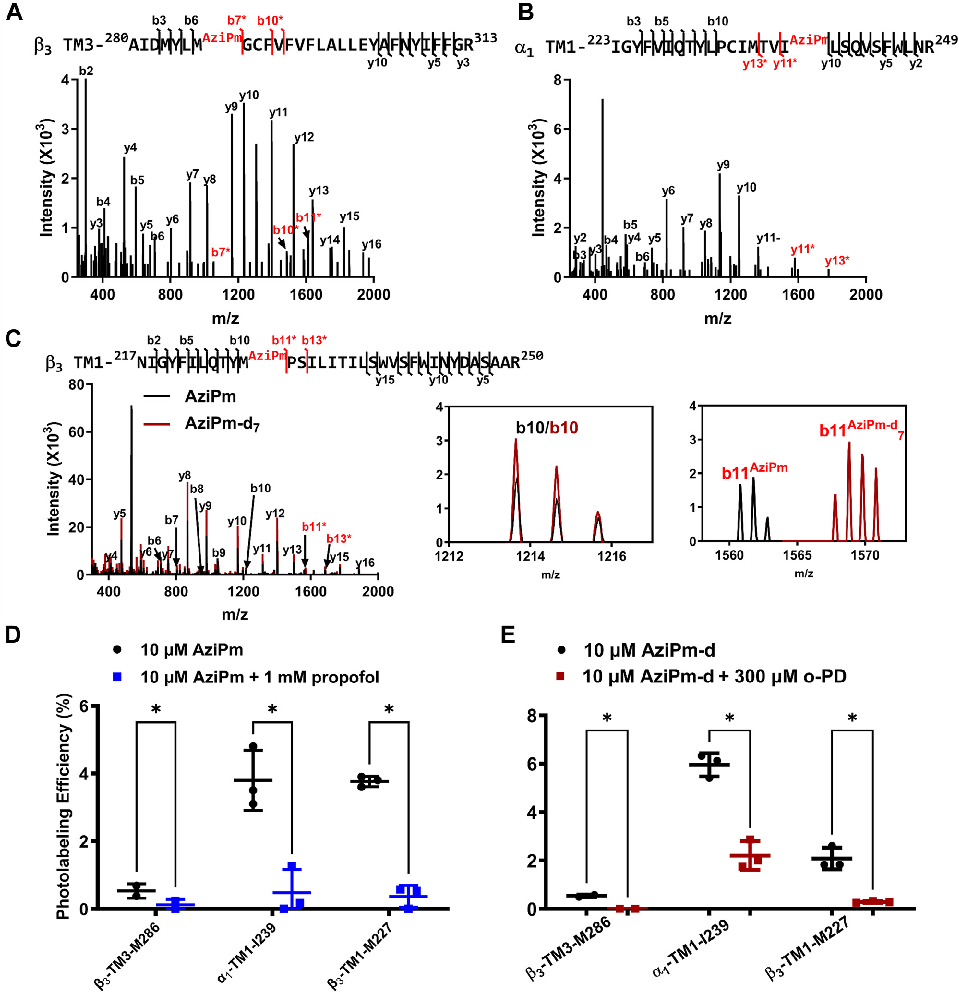
Figure 6
Structure-activity relationship analysis of propofol analogs at the different GABAA receptor binding sites, informing anesthetic drug design.
chart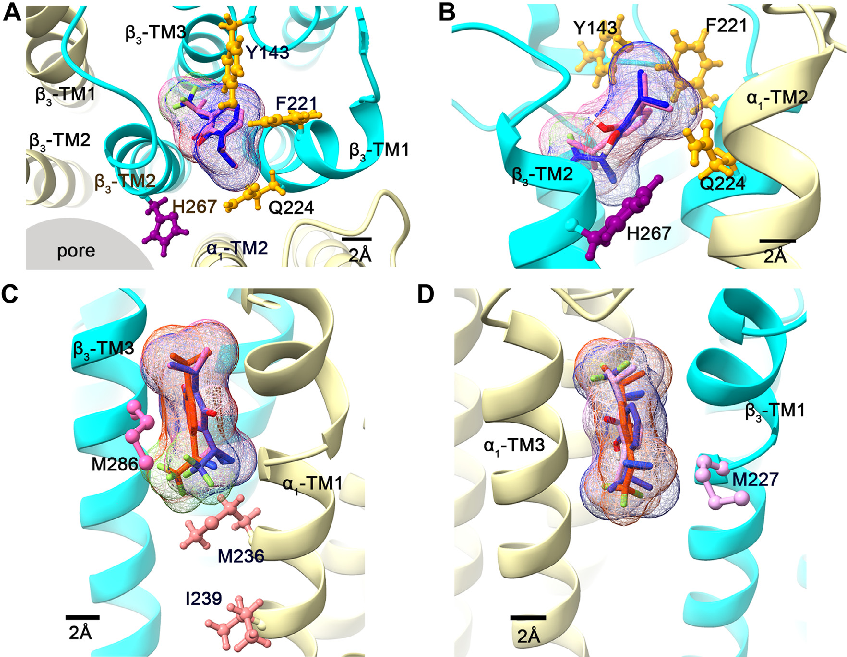
Figure 7
Molecular dynamics simulation or computational modeling of propofol interactions with GABAA receptor transmembrane domains.
chart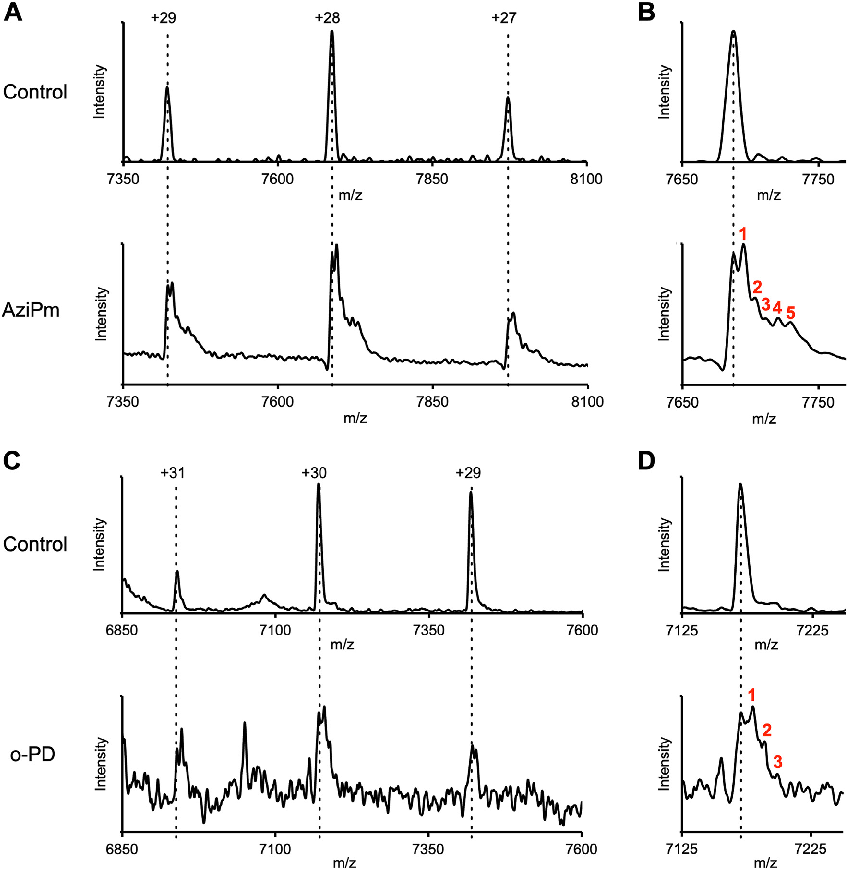
Figure 8
Comparative analysis of propofol binding site occupancy and functional consequences for GABAA receptor channel gating and anesthetic efficacy.
chart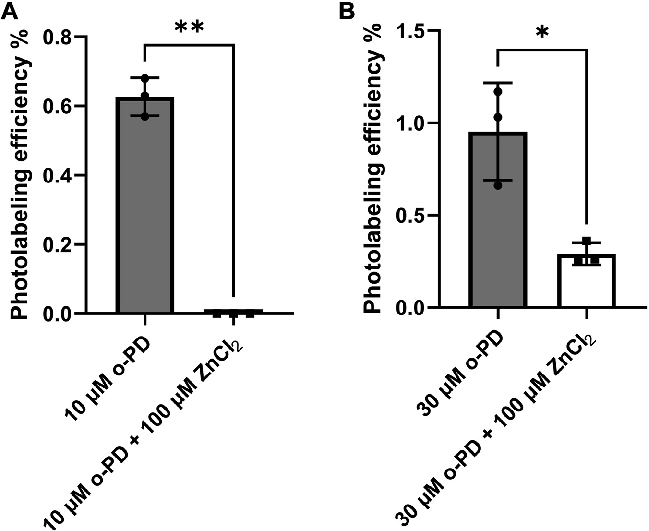
Figure 9
Supplementary structural or pharmacological data supporting the three-class model of propofol binding to GABAA receptors.
chart
Figure 10
Synthetic scheme for the preparation of cumene-d11 from benzene-d6, used as a deuterated propofol analog for binding site characterization studies on GABAA receptors.
diagramUsed In Evidence Reviews
Similar Papers
Pflugers Archiv : European journal of physiology · 2010
Glutamate receptors, neurotoxicity and neurodegeneration.
Oxidative medicine and cellular longevity · 2013
Cadmium and its neurotoxic effects.
Trends in neurosciences · 2001
Atypical neural messengers.
The Journal of nutrition · 2007
Metals and neurotoxicology.
Annals of hematology · 2018
Copper deficiency anemia: review article.
Neuroscience and biobehavioral reviews · 2017